Variant detection and grading overview
Developing pipelines for detecting variants from high throughput sequencing data is challenging due to rapidly changing algorithms and relatively low concordance between methods. This post will discuss automated methods providing evaluation of variant calls, enabling detailed diagnosis of discordant differences between multiple calling approaches. This allows us to:
- Characterize strengths and weaknesses of alignment, post-alignment preparation and calling methods.
- Automatically verify pipeline updates and installations to ensure variant calls recover expected variations. This extends the XPrize validation protocol to provide full summary metrics on concordance and discordance of variants.
- Make recommendations on best-practice approaches to use in sequencing studies requiring either exome or whole genome variant calling.
- Identify characteristics of genomic regions more likely to have discordant variants which require additional care when making biological conclusions based on calls, or lack of calls, in these regions.
This evaluation work is part of a larger community effort to better characterize variant calling methods. A key component of these evaluations is a well characterized set of reference variations for the NA12878 human HapMap genome, provided by NIST's Genome in a Bottle consortium. The diagnostic component of this work supplements emerging tools like GCAT (Genome Comparison and Analytic Testing), which provides a community platform for comparing and discussing calling approaches.
I'll show a 12 way comparison between 2 different aligners (novoalign and bwa mem), 2 different post-alignment preparation methods (GATK best practices and the Marth lab's gkno pipeline), and 3 different variant callers (GATK UnifiedGenotyper, GATK HaplotypeCaller, and FreeBayes). This allows comparison of openly available methods (bwa mem, gkno preparation, and FreeBayes) with those that require licensing (novoalign, GATK's variant callers). I'll also describe bcbio-nextgen, the fully automated open-source pipeline used for variant calling and evaluation, which allows others to easily bring this methodology into their own work and extend this analysis.
Aligner, post-alignment preparation and variant calling comparison
To compare methods, we called variants on a NA12878 exome dataset from EdgeBio's clinical pipeline and assessed them against the NIST Genome in a Bottle reference material. Discordant positions where the reference and evaluation variants differ fall into three different categories:
- Extra variants, called in the evaluation data but not in the reference. These are potential false positives.
- Missing variants, found in the NA12878 reference but not in the evaluation data set. These are potential false negatives. The use of high quality reference materials from NIST enables identification of genomic regions we fail to call in.
- Shared variants, called in both the evaluation and reference but differently represented. This could result from allele differences, such as heterozygote versus homozygote calls, or variant identification differences, such as indel start and end coordinates.
To further identify causes of discordance, we subdivide the missing and extra variants using annotations from the GEMINI variation framework:
- Low coverage: positions with limited read coverage (4 to 9 reads).
- Repetitive: regions identified by RepeatMasker.
- Error prone: variants falling in motifs found to induce sequencing errors.
We subdivide and restrict our comparisons to help identify sources of differences between methods indistinguishable when looking at total discordant counts. A critical subdivison is comparing SNPs and indels separately. With lower total counts of indels but higher error rates, each variant type needs independent visualization. Secondly, it's crucial to distinguish between discordance caused by a lack of coverage, and discordance caused by an actual difference in variant assessment. We evaluate only in callable regions with 4 or more reads. This low minimum cutoff provides a valuable evaluation of low coverage regions, which differ the most between alignment and calling methods.
I'll use this data to provide recommendations for alignment, post-alignment preparation and variant calling. In addition to these high level summaries, the full dataset and summary plots available below providing a starting place for digging further into the data.
Aligners
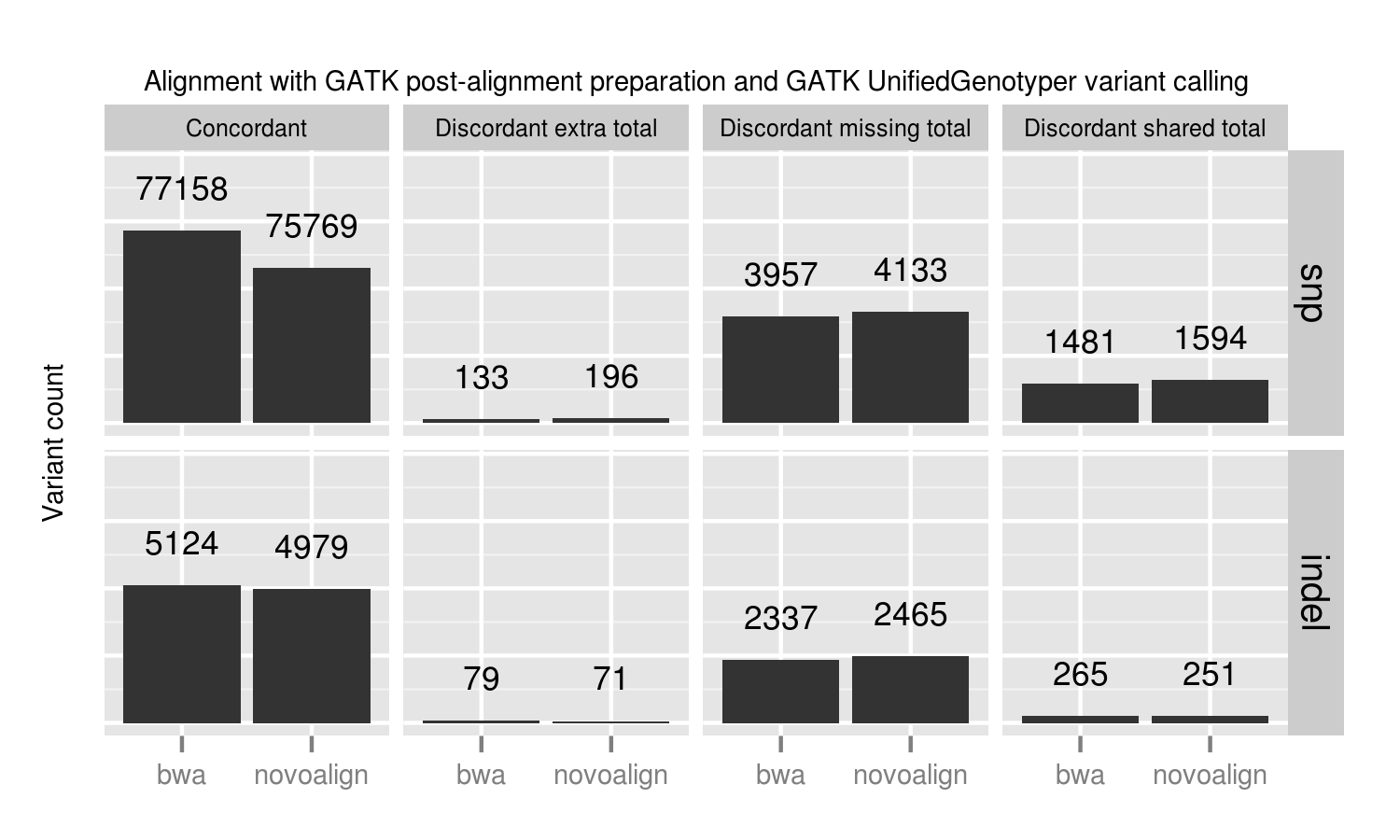
We compared two recently released aligners designed to work with longer reads coming from new sequencing technologies: novoalign (3.00.02) and bwa mem (0.7.3a). bwa mem identified 1389 additional concordant SNPs and 145 indels not seen with novoalign. 1024 of these missing variants are in regions where novoalign does not provide sufficient coverage for calling. Of those, 92% (941) have low coverage with less than 10 reads in the bwa alignments. Algorithmic changes impact low coverage regions more due to the decreased evidence and susceptibility to crossing calling coverage thresholds, so we need extra care and consideration of calls in these regions.
Our standard workflow uses novoalign based on its stringency in resolving large insertions and deletions. These results suggest equally good results using bwa mem, along with improved processing times. One caveat to these results is that some of the available Illumina call data that feeds into NIST's reference genomes comes from a bwa alignment, so some differences may reflect a bias towards bwa alignment heuristics. Using non-simulated reference data sets has the advantage of capturing real biological and process errors, but requires iterative improvement of the reference materials to avoid this type of potential algorithmic bias.
Post-alignment preparation and quality score recalibration
We compared two methods of quality recalibration:
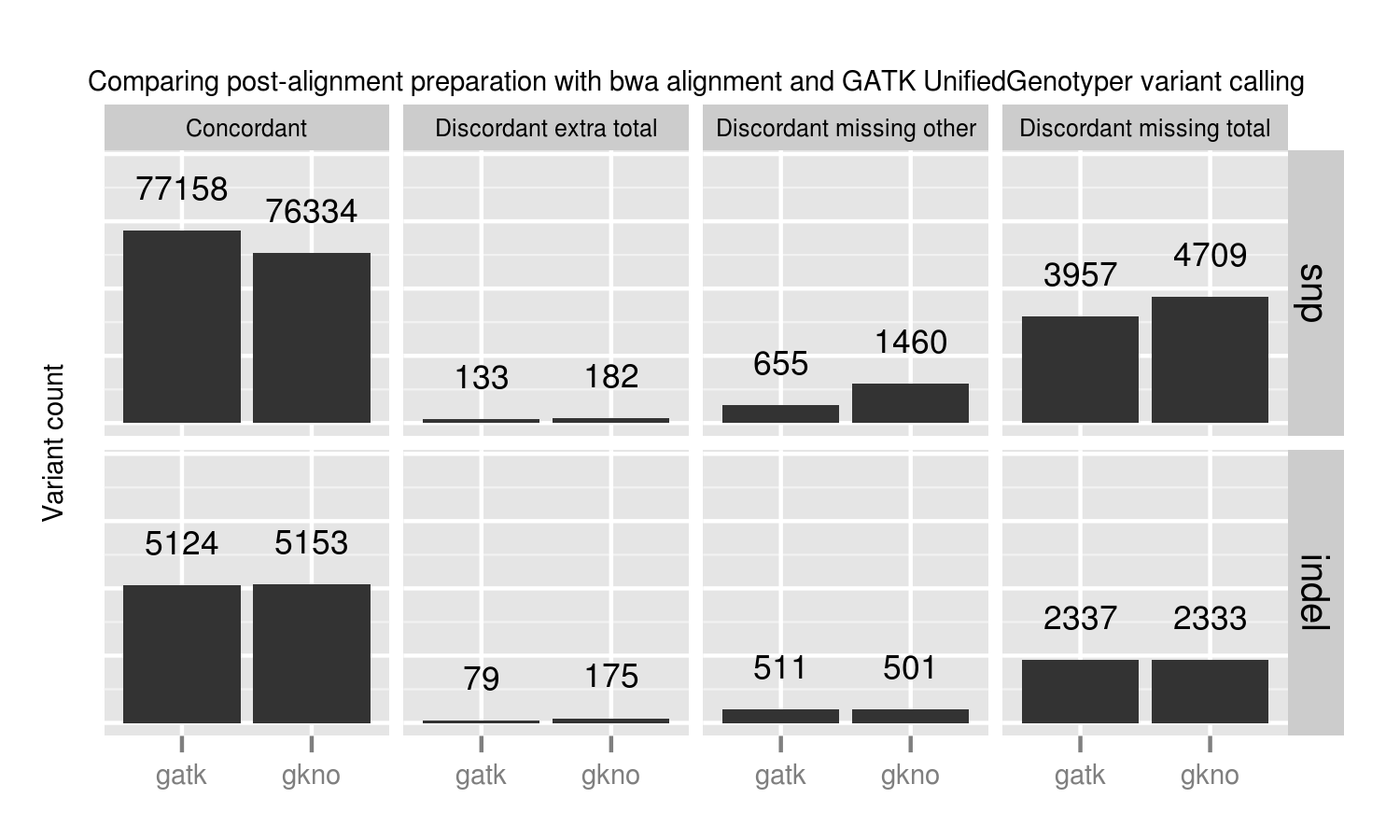
- GATK's best practices (2.4-9): This involves de-duplication with Picard MarkDuplicates, GATK base quality score recalibration and GATK realignment around indels.
- The Marth Lab's gkno realignment pipeline: This performs de-duplication with samtools rmdup and realignment around indels using ogap. All commands in this pipeline work on streaming input, avoiding disk IO penalties by using unix pipes. This piped approach improves scaling on large numbers of whole genome samples. Notably, our implementation of the pipeline does not use a base quality score recalibration step.
GATK best practice pipelines offer an advantage over the gkno-only pipeline primarily because of improvements in SNP calling from base quality recalibration. Specifically we lose ~1% (824 / 77158) of called variations. These fall into the discordant missing "other" category, so we cannot explain them by metrics such as coverage or genome difficulty. The simplest explanation is that initial poor quality calculations in those regions result in callers missing those variants. Base quality recalibration helps recover them. These results match Brendan O'Fallon's recent analysis of base quality score recalibration.
This places a practical number on the lost variants when avoiding recalibration either due to scaling or GATK licensing concerns. Some other options for recalibration include Novoalign's Quality Recalibration and University of Michigan's BamUtil recab, although we've not yet tested either in depth as potential supplements to improve calling in non-GATK pipelines.
Variant callers
For this comparison, we used three general purpose callers that handle SNPs and small indels, all of which have updated versions since our last comparison:
- FreeBayes (0.9.9 296a0fa): A haplotype-based Bayesian caller from the Marth Lab, with filtering on quality score and read depth.
- GATK UnifiedGenotyper (2.4-9): GATK's widely used Bayesian caller, using filtering recommendations for exome experiments from GATK's best practices.
- GATK HaplotypeCaller (2.4-9): GATK's more recently developed haplotype caller which provides local assembly around variant regions, using filtering recommendations for exomes from GATK's best practices.
Adjusting variant calling methods has the biggest impact on the final set of calls. Called SNPs differ by 4577 between the three compared approaches, in comparison with aligner and post-alignment preparation changes which resulted in a maximum difference of 1389 calls. This suggests that experimenting with variant calling approaches currently provides the most leverage to improve calls.
A majority of the SNP concordance differences between the three calling methods are in low coverage regions with between 4 and 9 reads. GATK UnifiedGenotyper performs the best in detecting SNPs in these low coverage regions. FreeBayes and GATK HaplotypeCaller both call more conservatively in these regions, generating more potential false negatives. FreeBayes had the fewest heterozygote/homozygote discrimination differences of the three callers.
For indels, FreeBayes and HaplotypeCaller both provide improved sensitivity compared to UnifiedGenotyper, with HaplotypeCaller identifying the most, especially in low coverage regions. In contrast to the SNP calling results, FreeBayes has more calls that match the expected indel but differ in whether a call is a heterozygote or homozygote.
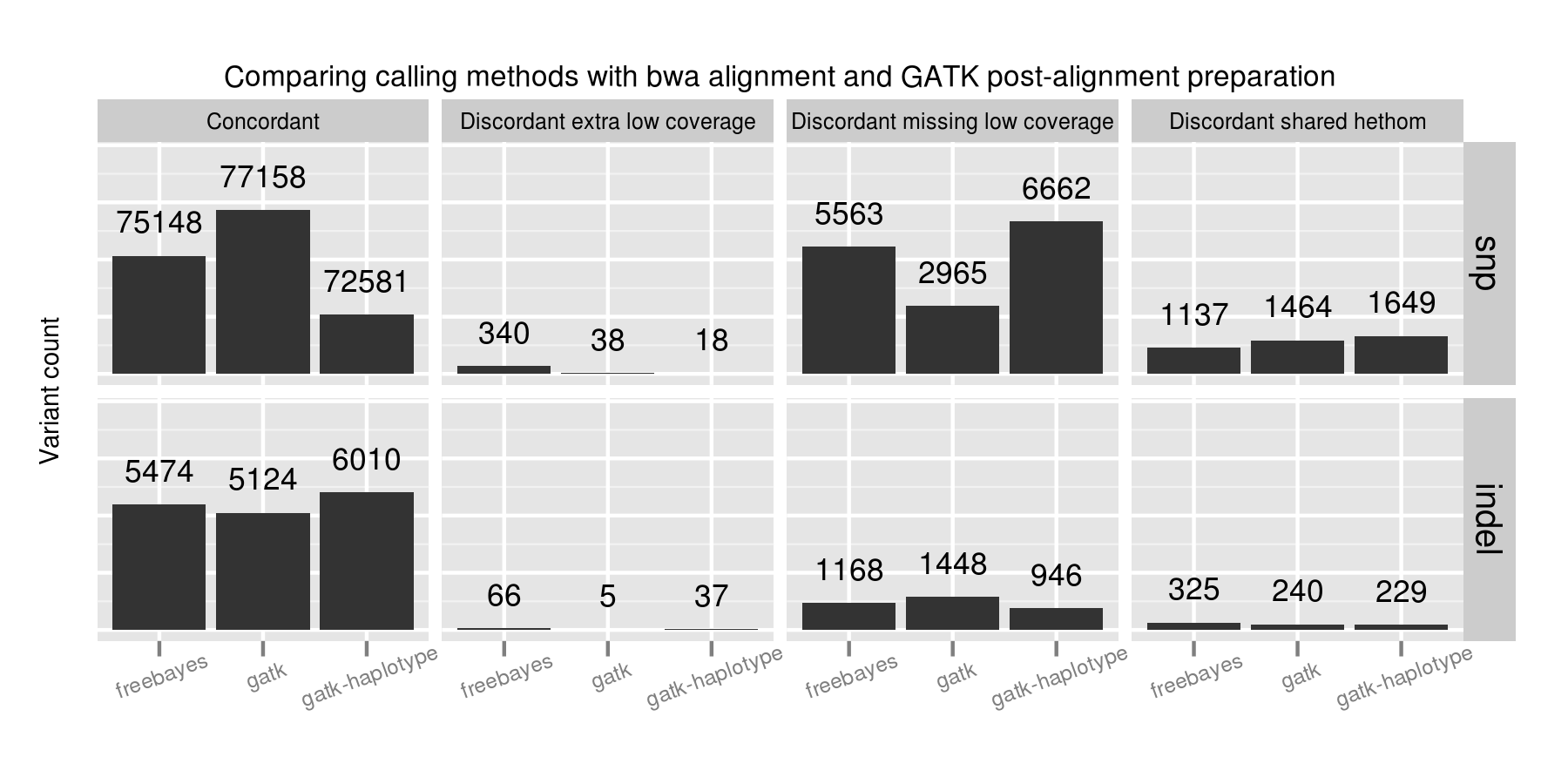
No one caller outperformed the others on all subsets of the data. GATK UnifiedGenotyper performs best on SNPs but is less sensitive in resolving indels. GATK HaplotypeCaller identifies the most indels, but is more conservative than the other callers on SNPs. FreeBayes provides intermediate sensitivity and specificity between the two for both SNPs and indels. A combined UnifiedGenotyper and HaplotypeCaller pipeline for SNPs and indels, respectively, would provide the best overall calling metrics based on this set of comparisons.
Low coverage regions are the key area of difference between callers. Coupled with the alignment results and investigation of variant changes resulting from quality score binning, this suggests we should be more critical in assessing both calls and coverage in these regions. Assessing coverage and potential false negatives is especially critical since we lack good tools to summarize and prioritize genomic regions that are potentially missed during sequencing. This also emphasizes the role of population-based calling to help resolve low coverage regions, since callers can use evidence from multiple samples to better estimate the likelihoods of low coverage calls.
Automated calling and grading pipeline
Method comparisons become dated quickly due to the continuous improvement in aligners and variant callers. While these recommendations are useful now, in 6 months there will be new releases with improved approaches. This rapid development cycle creates challenges for biologists hoping to derive meaning from variant results: do you stay locked on software versions whose trade offs you understand, or do you attempt to stay current and handle re-verifying results with every new release?
Our goal is to provide a community developed pipeline and comparison framework that ameliorates this continuous struggle to re-verify. The analysis done here is fully automated as part of the bcbio-nextgen analysis framework. This framework code provides full exposure and revision tracking of all parameters used in analyses. For example, the ngsalign module contains the command lines used for bwa mem and novoalign, as well as all other tools.
To install the pipeline, third-party software and required data files:
wget https://raw.github.com/chapmanb/bcbio-nextgen/master/scripts/bcbio_nextgen_install.py python bcbio_nextgen_install.py /usr/local /usr/local/share/bcbio-nextgen
The installer bootstraps all installation on a bare machine using the CloudBioLinux framework. More details and options are available in the installation documentation.
To re-run this analysis, retrieve the input data files and configuration as described in the bcbio-nextgen example documentation with:
$ mkdir config && cd config $ wget https://raw.github.com/chapmanb/bcbio-nextgen/master/config/\ examples/NA12878-exome-methodcmp.yaml $ cd .. && mkdir input && cd input $ wget https://dm.genomespace.org/datamanager/file/Home/EdgeBio/\ CLIA_Examples/NA12878-NGv3-LAB1360-A/NA12878-NGv3-LAB1360-A_1.fastq.gz $ wget https://dm.genomespace.org/datamanager/file/Home/EdgeBio/\ CLIA_Examples/NA12878-NGv3-LAB1360-A/NA12878-NGv3-LAB1360-A_2.fastq.gz $ wget https://s3.amazonaws.com/bcbio_nextgen/NA12878-nist-v2_13-NGv3-pass.vcf.gz $ wget https://s3.amazonaws.com/bcbio_nextgen/NA12878-nist-v2_13-NGv3-regions.bed.gz $ gunzip NA12878-nist-*.gz $ wget https://s3.amazonaws.com/bcbio_nextgen/NGv3.bed.gz $ gunzip NGv3.bed.gz
Then run the analysis, distributed on 8 local cores, with:
$ mkdir work && cd work $ bcbio_nextgen.py bcbio_system.yaml ../input ../config/NA12878-exome-methodcmp.yaml -n 8
The bcbio-nextgen documentation describes how to parallelize processing over multiple machines using cluster schedulers (LSF, SGE, Torque).
The pipeline and comparison framework are open-source and configurable for multiple aligners, preparation methods and callers. We invite anyone interested in this work to provide feedback and contributions.
Full data sets
We extracted the conclusions for alignment, post-alignment preparation and variant calling from analysis of the full dataset. The visualizations for the full data are not as pretty but we make them available for anyone interested in digging deeper:
- Summary CSV of comparisons split by methods and concordance/discordance types, easily importable into R or pandas for further analysis.
- Code for preparing and plotting results
- Full comparisons of all 12 methods, stratified by concordance and discordance: SNPs and indels
- Boxplots of differences between alignment methods: SNPs and indels
- Boxplots of differences between post-alignment preparation methods: SNPs and indels
- Boxplots of differences between variant calling methods: SNPs and indels
The comparison variant calls are also useful for pinpointing algorithmic differences between methods. Some useful subsets of variants:
- Concordant variants called by bwa and not novoalign, where novoalign did not have sufficient coverage in the region. These are calls where either novoalign fails to map some reads, or bwa maps too aggressively: VCF of bwa calls with low or no coverage in novoalign.
- Discordant variants called consistently by multiple calling methods. These are potential errors in the reference material, or consistently problematic calling regions for multiple algorithms. Of the 9004 shared discordants, the majority are potential false negatives not seen in the evaluation calls (7152; 79%). Another large portion is heterozygote/homozygote differences, which make up 1627 calls (18%). 6652 (74%) of the differences have low coverage in the exome evaluation, again reflecting the difficulties in calling in these regions. The VCF of discordants found in 2 or more callers contains these calls, with a 'GradeCat' INFO tag specifying the discordance category.
We encourage reanalysis and welcome suggestions for improving the presentation and conclusions in this post.