Small RNA-seq with bcbio-nextgen
The study of small RNA helps to understand part of the gene regulation of a cell. There are different types of small RNAs, the most important in mammals are miRNA, tRNA fragments and piRNAs. An advantage of small RNA-seq analysis is that we can study all small RNA types simultaneously, with the possibility to also detect novel small RNAs. bcbio-nextgen is a Python framework supported by a big scientific community that implements best practices for next-generation sequencing data and uses gold standard data to validate its analyses. It is well known for its variant calling and RNA-seq pipeline. Now bcbio has an small RNA-seq pipeline that allows quality control, adapter removal of fastq files, annotation of miRNA, isomiRs and tRNAs, and genome-wide characterization of other types of small RNAs. Here, I show the capabilities of the pipeline and its validation using data from the miRQC project.
miRQC project
miRQC provides samples with known relative amounts of small RNAs, enabling comparison of quantitation and detection of miRNAs. The main goal was to test different platforms for miRNA detection, but these are also great samples for benchmarking tools.
The samples in miRQC are: one Universal Human miRNA reference RNA (Agilent Technologies, #750700), one human brain total RNA (Life Technologies, #AM6050), several human liver total RNA (LifeTechnologies, #AM7960) and several MS2-phage RNA (Roche, #10165948001). Moreover, two more samples were created using different concentrations of UHmiRR and HBR. And finally, two miRNA families were spiked in human liver and MS2-phage samples.

Pipeline
There are 4 main steps in the small RNA-seq pipeline:
- adapter removal
- miRNA annotation
- de-novo miRNA detection
- other small RNAs detection
- quality control metrics
Adapter removal
We integrated cutadapt allowing a minimum read size of 17 nts and removing the adapter if it is at least 8 nts long.
miRNA annotation
bcbio uses
seqbuster for this
step. It has been used by
GEUVADIS consortium
for miRNA quantification and allows
isomiRs analysis as well. Read more
about why isomiRs are important.
Although not covered here, it is important to mention the pipeline uses miRDeep2 for de-novo miRNA discovery. In this case, it uses all samples for this analysis and then seqbuster for quantification of only the novel miRNAs.
Other smallRNAs detection
bcbio uses seqcluster to detect
unique units of transcription over the genome, allowing resolutions of small
RNAs found in multiple genomic locations. Normally
these small RNAs are dropped because they map multiple times on the genome and
require special analysis to avoid bias in the quantification. Read more about
why
other small RNAs are important.
This tool produce a sqlite3 database for visualization. An image example is here.
Quality control metrics
bcbio summarizes Fastqc metrics for each sample. Together with different
metrics from the previous steps, the user has an idea of the quality of the
samples and the overall project. It includes fastqc results, size
distribution after adapter removal and amount of small RNAs mapped to miRNAs,
tRNA, rRNA, repeats among others. Other metrics such as, amount of data used until
the end of the analysis, or warning flags if the data is noisy, are provided by
seqcluster and included in the final R markdown template report.
Reporting
bcbio generates a
R markdown template report
to visualize results from each of the steps. It
is inside the report folder in the working directory or final folder after the analysis.
Results
The mirRQC samples allow us to measure quantitation and detection
accuracy of specific miRs for the tools integrated in bcbio.
Size distribution
The size distribution is a first estimate of the quality of your data. In a normal small RNA sample we should see a peak at 22/23nt and maybe another at 26 or 31 depending on the biology of the samples.

note: there may be specific cases where this assumption is not true, but it applies to the majority of projects.
miRNA abundance detection of miRQC samples
There are 4 samples that help to validate the quantification analysis:
- A: universal human RNA sample
- B: human brain sample
- C: 25% of A + 75% of B
- D: 25% of B + 75% of A
So we can assume:
- If A > B then A > D > C > B
- If B > A then A < D < C < B
Note that C/D samples are swapped in the paper and in the GEO web. The text from the paper says:
These samples (termed miRQC A–D) consist of 100% Universal Human miRNA Reference RNA (UHmiRR; A), 100% human brain RNA (HBR; B) and two titrations thereof (C = 0.75A + 0.25B and D = 0.25A + 0.75B).
While the text in the GEO web says:
Source name miRQC C Organism Homo sapiens Characteristics biomaterial: Mixture of 25% UHRR and 75% HuBr Total RNA
Source name miRQC D Organism Homo sapiens Characteristics biomaterial: Mixture of 75% UHRR and 25% HuBr Total RNA
After the analysis with bcbio, we can calculate the miRs that
follow the relative abundance rule. To measure this, I took the average from
the replicates and kept only the miRs with a median > 5 counts after
upper quantile normalization among samples.
miRNAs which A > B are 111, and all of them follows A > D > C
miRNAs which B > A are 181 and 174 follows B > C > D
That is more than 95% of accuracy for miRs with more than 5 counts.
Specificity
To evaluate specificity we used samples that included specific miRNAs that are not normally expressed there. These samples were analyzed in a different run.
This analysis uses samples from 4 human liver RNAs and 4 MS2-phage RNAs with 8 synthetic miRNAs from two families (miR-302a/b/c/d and let-7a/b/c/d).
We should only see those miRNAs (let-7 and miR-302 families) in those samples. Only sequences that appear > 2 times in the fastq files are considered.
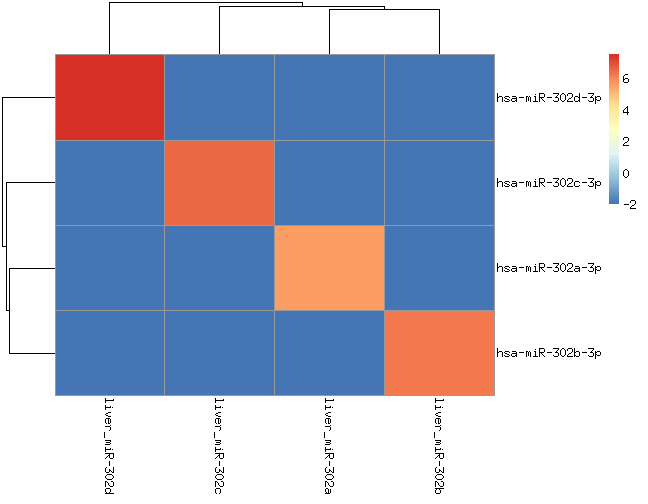
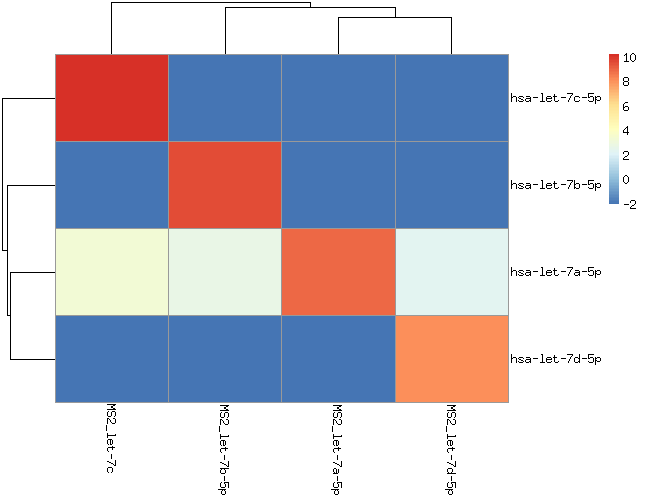
hsa-let-7a appears in all samples. If you check the processed data, this is not due to incorrect alignments. It would be interesting to figure out whether this signal is because of contamintation or errors during sequencing or/and during amplification.
Cluster abundance detection of miRQC samples
Similarly to miRs, we can validate other small RNAs detected by
seqcluster. The results were very similar:
- clusters which A > B are 147, where 139 (75 are known miRNAs) follow D > C
- clusters which B > A are 230, where 222 (129 are known miRNAs) follow D > C
Timing and Resources
The running time for 8 samples with 6 millions reads each was 3 hour and 19 minutes.
| Total | 3:19 | total cores | total memory GB |
|---|---|---|---|
| organize samples | 0 | 1 | 1 |
| trimming & miRNA | 0:21 | 8 | 20 |
| prepare | 0:01 | 1 | 8 |
| alignment | 0:07 | 6 | 42.1 |
| cluster | 2:49 | 1 | 8 |
| quality control | 0:01 | 8 | 20 |
| report | 0 | 1 | 1 |
Conclusion
We conclude that the current analysis has a reliable quantification and specificity of miRNAs and other small RNA molecules. What’s more, it helps with the downstream analysis creating a complete R markdown template that covers the most important section of small RNA studies.
I am currently implementing post-processing steps of the tDRmapper (analysis of tRNAs) output to allow an easy differential expression or clustering in downstream analysis. In the future, I would like to implement proTRAC for the analysis of piRNAs.
Thanks
- Harvard T.H. Chan School of Public Health for supporting the integration of small RNAseq pipeline in bcbio. Special thanks to @roryk and @chapmanb.
- Research Computing at Harvard Medical School: Chris Botka, Director of Research Computing and all the people in the team.
- Special thanks to the authors for making their validation data available. I encourage use of this data for any tool that analyzes small RNAs.